Abstract:
The Cu-terpyridine complex was studied through quantum chemical
calculations using B3LYP method with 6311+G(d,p) basis sets in
the GAUSSIAN 2003 program package. Copper atoms and ions can
bond to terpy in a mono-, bi- or tridentate mode. The geometry
optimizations were carried out for the mono-, bi-, and
tridentate structures of the Cu-terpy complex. The geometry
optimizations yielded five local minimum energy structures: a
tridentate (T), a bidentate (B), and three monodentate isomers
(MI, MII and MIII). The lowest
energy structure was the T isomer and the second lowest energy
structure corresponds to the B isomer and was located ~ 6000 cm-1
higher in electronic energy than the T isomer. The MI,
MII and MIII isomers were separated by
about 1000 cm-1, with the most stable M conformer
being about 2000 cm-1 higher in electronic energy
than the B form. Due to the different numbers of Cu-N σ-bonds in
the monodentate, bidentate, and tridentate forms, the predicated
BDEs were in the order of
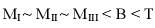
for both the neutral and ionic isomers of Cu-terpy complex.
Keywords: Copper, terpyridine, computational calculations,
metal-ligand bonding.